Neurosciences/Le métabolisme cérébral
Le cerveau est un organe qui consomme beaucoup d'énergie : près de 20% de la consommation énergétique du corps est de son fait, alors qu'il ne représente que 2 % de sa masse. Il faut dire que faire fonctionner les neurones demande de l'énergie, d'autant plus que ceux-ci sont actifs. Générer des potentiels d'action demande une certaine énergie, produire des neurotransmetteurs aussi, sans compter l'action des pompes et canaux ioniques de la membrane. Le cerveau utilise la majeure partie de son énergie pour faire fonctionner ses pompes et canaux ioniques, principalement les pompes. Celles-ci vont, pour rappel, pomper certains ions en dehors de la cellule, contre leur gradient de concentration. Cela demande naturellement de l'énergie, pour contrer l'effet de la concentration plus importante dans le milieu extérieur. Chaque pompe va ainsi utiliser une ou plusieurs molécules d'ATP pour faire sortir un ion. Cela ne parait n'être pas grand-chose, mais cela compte pour 50% du métabolisme de base du cerveau !
Le métabolisme énergétique du cerveau
Le cerveau est un organe qui consomme beaucoup d'énergie. On estime qu'il est responsable, à lui seul d'environ 25 % de la consommation énergétique du corps. La valeur exacte varie beaucoup selon beaucoup de paramètres. Par exemple, on sait que la consommation énergétique du cerveau dépend de sa température. On estime qu'une réduction de 1°C réduit la consommation énergétique cérébrale de 6 à 5 %. Cela a d'ailleurs des applications thérapeutiques, dans les cas d'AVC, d'arrêt cardiaque ou d'autres situations similaires où le cerveau manque d'oxygène. Dans ces situations, on refroidit le cerveau pour ralentir son métabolisme. Cela réduit l'apparition de lésions cérébrales, causées par un métabolisme anormal lié au manque d'oxygène. Quoi qu’il en soit, le métabolisme cérébral dépend de bien d'autres paramètres, et il serait inutile d'en faire une liste exhaustive.
Les sources d'énergie du cerveau
Les neurones sont comme toutes les cellules : ils consomment des nutriments pour fabriquer de l'énergie, qu'ils emmagasinent sous forme d'ATP dans la cellule. En temps normal, ces nutriments sont des sucres, mais le cerveau peut aussi bruler des dérivés de la désagrégation des graisses ou des protéines si le sucre vient à manquer. Cette combustion des nutriments, implique toute une série de réactions chimiques très compliquées. Ceux qui s'y connaissent en biologie devraient connaitre les notions de fermentation ou de respiration cellulaires, voire le cycle de Krebs. Nous n'allons pas revenir sur ces notions fondamentales de la biologie, mais nous allons remarquer que la combustion des nutriments peut se faire soit en présence d'oxygène, soit sans. Quand les nutriments sont brulés en présence d'oxygène, les réactions chimiques sont des réactions dites de respiration cellulaire. Dans le cas contraire, ce sont des réactions de fermentation. Les neurones sont capables à la fois de respiration cellulaire que de fermentation. Vu que les neurones consomment beaucoup d'énergie, le processus principal est la respiration, ce qui fait que le cerveau a besoin d'oxygène pour fonctionner. Pour résumer, le cerveau a principalement besoin de sucres et d'oxygène, mais il peut consommer des graisses en cas de manque.
Le glucose et l'oxygène
En temps normal, le cerveau consomme du sucre par respiration cellulaire, la fermentation étant minimale. Le sucre le plus utilisé par le cerveau est le glucose, du moins en temps normal, mais des molécules similaires au glucose peuvent être utilisées en lieu de place de celui-ci : c'est le cas pour le pyruvate, le mannose et le lactate. Les expériences qui ont montré cela sont assez simples à expliquer. Elles se bornent à comparer la composition du sang entre les artères qui alimentent le cerveau et les veines qui en sortent. Ces mesures montrent alors que le sang est identique entre l'entrée et la sortie, si ce n'est que sa teneur en glucose et en oxygène est plus basse dans les veines que dans les artères, sans compter que sa teneur en dioxyde de carbone augmente.
Les mesures précédentes traduisent le fait que le cerveau utilise ce qu'on appelle la respiration cellulaire aérobie pour produire son énergie : il consomme de l'oxygène et du glucose et produit de l'énergie et du dioxyde de carbone. Cependant, ces mesures montrent que tout l'oxygène n'est pas converti de cette manière. Si tout le glucose était utilisé ainsi, 100 grammes de cerveau devraient consommer 26 millimoles de glucose par minutes. Or, la valeur mesurée est de 31 millimoles par minutes. Il y a donc une petite différence de 4,4 millimoles par minutes, qui est utilisée autrement. Dans le détail, on verra que ce glucose est transformé et stocké dans les astrocytes et les neurones, il est mis en réserve.
Les lipides et acides gras
Outre le glucose, les protéines et des dérivés de la désagrégation des graisses peuvent être consommés par le cerveau pour produire de l'énergie. Encore une fois, cette production s'effectue par le biais de la respiration cellulaire aérobie et le cerveau n'est pas le seul à pouvoir faire cela (tous les tissus peuvent brûler des protéines ou des lipides dans le cycle de Krebs). Par contre, le métabolisme cérébral des graisses est différent du métabolisme des autres tissus. En effet, le cerveau ne peut pas bruler les acides gras pour produire de l'énergie, contrairement aux autres tissus. La raison est que les acides gras ne peuvent pas traverser la barrière hémato-encéphalique. En revanche, le cerveau peut bruler les corps cétoniques, des dérivés de la désagrégation des graisses qui peuvent traverser la barrière hémato-encéphalique et alimenter le cerveau en énergie. Parmi les corps cétoniques, deux alimentent le cerveau et le cœur en énergie : l'acétylacétate et le β-D-hydroxybutyrate.
Cependant, la production de corps cétoniques n'arrive que dans des conditions très particulières. Il faut que le corps manque de sucres (glucose) au point que le foie en est réduit à bruler des graisses. Le métabolisme des lipides et acides gras donne alors naissance à des corps cétoniques, par un processus dit de cétogenèse. Par exemple, cela arrive en cas de diabète ou de jeûne : l'alimentation en sucre étant alors inadéquate, le cerveau doit utiliser d'autres voies que la consommation de glucose. Un autre exemple est celui des nouveaux-nés allaités, dont l'alimentation est très riche en lipides et en acides gras. Dans tous les exemples précédents, les réactions chimiques utilisées pour produire de l'énergie à partir de lipides ou de protéines sont les mêmes : les voies métaboliques des nouveaux-nés sont réactivées en cas de jeûne ou de diabète.
L'hypoxie cérébrale
Quand l'oxygène vient à manquer, on fait face à une situation d'hypoxie cérébrale. Une telle situation arrive notamment lors d'un AVC ou d'un arrêt cardiaque, quand le cerveau n'est plus irrigué par le sang. L'oxygène n'est plus apporté au cerveau, qui réagit pour faire face à la pénurie. Le cerveau a beau avoir une grosse consommation énergétique, il a des réserves très faibles en nutriments. Ce qui fait que l'hypoxie commence à causer des problèmes en quelques minutes, voire quelques secondes. Il se produit alors une cascade de réactions chimiques, qui endommage les neurones et entraine leur mort. Cette cascade se produit en deux étapes : une première étape liée à la carence en oxygène, une autre quand le sang revient dans le cerveau et où l'oxygène revient. Ces deux étapes laissent des dommages cérébraux massifs, qu'il s'agisse du manque d'oxygène ou des dommages de la seconde étape, appelés dommages de re-perfusion.
La cascade ischémique
La première étape est une série de réactions chimiques qui s'appelle la cascade ischémique, qui fait suite à un manque d'oxygène. Pour faire simple, le cerveau réagit en passant à des réactions de fermentation pour consommer les sucres. Mais l'usage de la fermentation pose divers problèmes. En premier lieu, elle ne fournit pas assez d'énergie pour les pompes et canaux ioniques. L'équilibre ionique du cerveau est alors perturbé, avec une accumulation d'ions dans ou en dehors des neurones. En second lieu, la fermentation produit des déchets qui sont toxiques pour les neurones. Or, ceux-ci peuvent entrainer des dommages s'ils ne sont pas évacués du cerveau. La majorité des dommages provient, de manière indirecte, du déséquilibre ionique, qui intoxique les neurones de l'intérieur.
Le dysfonctionnement principal est l'arrêt de certaines pompes ioniques, notamment des pompes au potassium, au calcium et au sodium. Cela mène à une accumulation de sodium et de calcium dans les neurones. En premier lieu, le calcium va s'accumuler dans les neurones. Or, le calcium est toxique pour les cellules, ce qui peut forcer l'apoptose des cellules (leur suicide). De plus, ce calcium favorise la libération des neurotransmetteurs, dont le glutamate. Or, le glutamate fait rentrer encore plus de calcium via les récepteurs NMDA et AMPA. Rappelons que c'est pour cela que le glutamate a des propriétés excitotxiques, à savoir qu'il peut exciter les neurones à l'excès, jusqu’à en devenir toxique. Les effets excitotoxiques du glutamate proviennent de là, et ceux-ci s'expriment avec force lors de l'ischémie, bien plus que dans des conditions normales. Limiter les dégâts de la cascade ischémique demande d'agir vite afin de stopper celle-ci avant qu'un trop grand nombre de neurones soient morts. Une autre possibilité serait d'atténuer l'excitotoxicité du glutamate, en utilisant des antagonistes des récepteurs NMDA et AMPA. Mais cette stratégie n'a pas donné de bons résultats dans les études réalisées à l'heure actuelle (mi-2017).
Un autre défaut est que le sodium s'accumule dans les neurones, ce qui en perturbe l'équilibre hydroélectrique (pour les connaisseurs, l'équilibre osmotique). Les raisons à cela sont multiples. Déjà, les pompes ioniques calcium-sodium vont tenter d'évacuer le calcium dans les neurones, mais cela fait rentrer du sodium. De plus, certaines pompes ioniques potassium-sodium vont dysfonctionner, en raison du manque d'ATP, ce qui réduit l'élimination de sodium intraneuronale. Tout cela fait que les neurones tendent à accumuler beaucoup d'ions sodium dans leur cytoplasme. Sans rentrer dans les détails, cela a une conséquence : de l'eau s'accumule dans les neurones, qui gonflent. Il se produit alors un œdème cérébral, qui fait gonfler le cerveau et le compresse sur le crâne. Cette pression entraine des lésions assez graves, si elle est assez intense. Mais nous en reparlerons dans le chapitre sur la pression intracrânienne, qui parlera des œdèmes cérébraux en général.
Enfin, la fermentation produit des déchets métaboliques, les deux principaux étant : le lactate et des ions hydrogène. Les deux sont toxiques pour les neurones, quand ils sont en excès, ce qui est le cas lors d'une hypoxie cérébrale. L'accumulation de lactate est moins problématique que l'accumulation des ions hydrogène. Cette dernière fait que le pH diminue, entraînant une acidose métabolique, perturbant les réactions chimiques cérébrales.

Les interactions neurones-astrocytes
Les molécules dissoutes dans le sang doivent traverser la barrière hémato-encéphalique pour arriver aux neurones. Leur passage à travers celle-ci fait intervenir des molécules appelées transporteurs. Pour rappel, ce sont des "récepteurs" qui permettent à une molécule de passer d'un côté à l'autre d'une membrane cellulaire. Ici, la molécule en question est une molécule sanguine et les membranes sont celles de la barrière hémato-encéphalique. Les transporteurs sont spécialisés, dans le sens où ils ne laissent passer que quelques molécules bien précises et pas les autres. Par exemple, les transporteurs pour le glucose permettent uniquement le passage du glucose, mais pas des autres molécules. Quoi qu’il en soit, les molécules sanguines traversent la barrière hémato-encéphalique, du moins pour celles qui ont un transporteur adapté, mais elles n'arrivent pas directement dans les neurones ou dans le fluide entre les neurones. Rappelons que la barrière hémato-encéphalique est composée de deux couches : un vaisseau sanguin, entouré par des astrocytes. Les transporteurs localisés à la surface des vaisseaux sanguins cérébraux permettent aux molécules de passer dans les astrocytes, qui se chargent ensuite de les redistribuer aux neurones par divers mécanismes. Pour résumer, les molécules traversent la barrière hémato-encéphalique, se retrouvent dans les astrocytes, puis sont transférées aux neurones à la demande. Autant dire que les interactions entre neurones et astrocytes sont très importantes pour le métabolisme cérébral.
Rappelons que les astrocytes n'ont pas beaucoup de transporteurs différents, les principaux étant les transporteurs pour le glucose. L'absence de transporteurs pour de nombreuses molécules/ions empêche la majorité des ions et molécules de traverser les astrocytes et d'atteindre le cerveau. C'est pour cela que la barrière hémato-encéphalique est aussi sélective.
Le métabolisme du glucose
Les astrocytes ont un rôle très important dans le métabolisme énergétique des neurones. Pour simplifier, les astrocytes servent de réservoir d'énergie à disposition des neurones : ils captent le glucose sanguin, le mettent en réserve, et le distribuent aux neurones à la demande. Ils servent donc de réservoir d'énergie, dans lequel les neurones alentours peuvent puiser s'ils en ont besoin. À noter que les astrocytes stockent l'énergie non pas sous la forme de glucose, mais dans des molécules de glycogène et de lactate. Rappelons que le glycogène est synthétisé à partir du glucose. Le glycogène se fabrique en liant plusieurs molécules de glucose entre elles, d'une manière extrêmement compacte. C'est la forme sous laquelle les sucres sont stockés dans la plupart des cellules, cette molécule contenant une grande quantité d'énergie dans un volume très petit. Les astrocytes en concentrent de grandes quantités pour répondre aux besoins métaboliques des neurones, ainsi que pour leur fonctionnement. Si le besoin s'en fait sentir, les liaisons de la molécule de glycogène sont brisées par des enzymes, ce qui libère de nombreuses molécules de glucose, de lactate, ou d'autres formes de sucres.
Le transfert du glucose des astrocytes aux neurones peut se faire de diverses manières, mais il se fait principalement sous la forme de lactate. Les astrocytes libèrent du lactate dans le milieu extracellulaire, qui est capté par les neurones. Ce lactate est produit dans les astrocytes par dégradation du glucose et est destiné non au stockage, mais à la consommation immédiate. Dans le détail, le glucose est dégradé en pyruvate, qui est lui-même transformé en lactate. Le lactate est alors envoyé aux neurones, qui synthétisent du pyruvate avec, pyruvate qui est utilisé par la respiration aérobie (cycle de Krebs). On peut préciser que la libération du lactate par les astrocytes est couplée aux besoins des neurones par divers mécanismes. Ce qui veut dire que les astrocytes détectent que les neurones ont besoin d'énergie, et libèrent du lactate selon quand les besoins s'en font sentir. Le premier est que les neurones ont surtout besoin d'énergie, et donc de sucres, après avoir émis des potentiels d'action. Or, les astrocytes mesurent en permanence la quantité de neurotransmetteurs dans le milieu extra-cellulaire : plus il y en a, plus les neurones ont dépensés d'énergie pour les émettre et plus ils ont besoin d'énergie. Pour être plus précis, ils mesurent la quantité de glutamate, non de tous les neurotransmetteurs. Pour résumer, la liaison du glutamate sur les récepteurs astrocytaires va stimuler la libération du lactate dans le milieu extra-cellulaire, qui est ensuite assimilé par les neurones.
Évidemment, le métabolisme du glucose implique l'existence de transporteurs du glucose à la surface des astrocytes et des neurones. Dans l'ensemble des tissus, il existe quatorze transporteurs du glucose différents, qui n'ont pas la même composition chimique et sont de forme différente. Ils sont appelés GLUT-1, GLUT-2, GLUT-3, ..., GLUT-14, GLUT étant l'abréviation de GLUcose-Transporter. Ils ne sont pas spécifiques du glucose, mais peuvent aussi servir de transporteurs à d'autres "sucres". Par exemple, le GLUT-5 sert de transporteur pour le glucose, mais aussi le fructose. D'autres servent de transporteur pour l'urate, le myoinositol et quelques autres molécules. Toujours est-il que l'on peut classer ces transporteurs du glucose en trois grandes catégories, mais seuls ceux de la première sont présents dans le cerveau, à savoir les récepteurs de GLUT-1 à GLUT-4 et le GLUT-14. Le transporteur GLUT1 est surtout présent dans la membrane des astrocytes, de même que le transporteur GLUT2. Le premier est présent dans tous les astrocytes cérébraux, alors que GLUT2 est présent dans certaines astrocytes, localisés dans des aires cérébrales bien précises (hypothalamus et tronc cérébral). Le transporteur GLUT3 est la chasse gardée des neurones.
Le métabolisme du cholestérol
Le cholestérol est un lipide intervenant dans de nombreuses fonctions métaboliques du corps, et joue notamment un rôle dans le fonctionnement du cerveau. Il s'agit en effet d'un des composants principaux de la myéline qui entoure les axones (la gaine de myéline). Sachant que plus de la moitié du cerveau est composée de substance blanche, donc de myéline, il n'est pas étonnant que le cerveau soit l'organe le plus riche de tous en cholestérol. Certaines études estiment que près de 20% du cholestérol total est localisé dans le cerveau. De plus, le cholestérol est un composant de la membrane des neurones et des cellules gliales. Plus les axones et dendrites d'un neurone se développent, plus ceux-ci doivent fabriquer de membrane cellulaire pour donner naissance aux axones et dendrites. Pour résumer, les besoins en cholestérol augmentent avec l'apparition de nouvelles synapses et dendrites.
La quantité de cholestérol cérébral reste approximativement constante au cours du temps. Ce n'est que lors de myélinisation du système nerveux que le cholestérol est fortement synthétisé. Lors de ces périodes, la formation des gaines de myéline augmente les besoins en cholestérol, qui ne peuvent être assouvis que par une synthèse importante. Les périodes de développement des synapses/neurones entrainent une forte consommation en cholestérol, les besoins étant alors fortement augmentés. En-dehors de ces périodes où les besoins sont augmentés, la synthèse de cholestérol cérébral est contrebalancée par sa consommation et son élimination. Le métabolisme cérébral du cholestérol est illustré dans le schéma ci-dessous, que je vous invite à regarder avant de lire ce qui va suivre.

Le cholestérol sanguin n'est pas la source de cholestérol cérébral, vu que le cholestérol sanguin ne traverse pas la barrière hémato-encéphalique. Le cholestérol cérébral est synthétisé par les astrocytes, encore eux, sous la forme d'apolipoprotéines (ApoE). Ces apolipoprotéines sont synthétisées sous une forme immature, avant d'être finalisées par l'action de protéines membranaires de type ABCA. Elles sont ensuite absorbées par les neurones et sont transformées en cholestérol à l'intérieur des neurones, via l'action d'une enzyme.
Le cholestérol des neurones va ensuite être utilisé par le neurone. Il servira notamment dans la fabrication de la membrane du neurone. Rappelons que le neurone ne produit pas sa gaine de myéline, mais que ce sont les oligodendrocytes qui le font, ce qui fait que le choléstérol capté par les neurones ne sert pas à fabriquer la gaine de myéline. Mais l'utilisation principale du cholestérol intra-neuronal est de loin la formation de protéines appelées bêta-amyloides, présentes à la surface des cellules. On peut signaler que cette protéine est impliquée dans la maladie d'Alzheimer, bien que l'on sache mal comment... Rappelons que le cholestérol cérébral est aussi impliqué dans la fabrication de neurostéroïdes, des stéroïdes qui modulent l'action du GABA et du glutamate. Leur fabrication demande une conversion du cholestérol en prégnénolone, qui est elle-même transformée en divers stéroïdes, puis en neurostéroïdes. La conversion du cholestérol en prégnénolone est effectuée par l'enzyme CYP450scc et a lieu dans les mitochondries. Pour cela, le cholestérol est capté un transporteur qui capte le cholestérol cytoplasmique et le fait rentrer dans les mitochondries, à l'intérieur desquelles il s'effectue la transformation.
Il arrive que le cerveau produise un peu plus de cholestérol que demandé. Dans ce cas, le cholestérol produit en excès (non-utilisé par le cerveau) est soit stocké, soit éliminépar diverses voies métaboliques. Seul 1% du cholestérol cérébral est stocké dans les neurones et astrocytes. Il y prend alors la forme de gouttelettes de graisses de petite taille. Ce stockage demande que le cholestérol subisse des modifications chimiques, induites par une enzyme appelée cholesterol acyltransferase 1 (ACAT1/SOAT1).
Les neurones qui contiennent l'enzyme CYP46 peuvent dégrader le cholestérol en excès en une molécule appelée cérébrostérol. Appellation bien plus facile à retenir que le nom scientifique du cérébrostérol, à savoir : 24S-hydroxycholestérol. Ce cérébrostérol est émis dans le milieu-extracellulaire où il est soit éliminé, soit recyclé par les astrocytes. Le cérébrostérol peut traverser la barrière hémato-encéphalique, ce qui lui permet d'être éliminé dans la circulation sanguine. Plus de 2/3 du cérébrostérol est éliminé, le reste étant capté par les astrocytes et recyclé en ApoE.
Le métabolisme des neurotransmetteurs
Les astrocytes ne vont pas seulement fournir des nutriments aux neurones : ils vont aussi jouer un grand rôle dans le recyclage de certains neurotransmetteurs, comme le GABA, le glutamate, la glycine, et autres. Dans les faits, ces neurotransmetteurs sont produits à partir respectivement de sérine (glycine) et de glutamine (glutamate et GABA).
Prenons le cas de la sérine et de la glycine comme premier exemple. La sérine est produite dans les astrocytes à partir du glucose, avant d'être envoyé aux neurones. Les neurones vont alors synthétiser de la glycine à partir de cette sérine et l'utiliser comme neurotransmetteur. Après utilisation, la glycine est recapturée par les astrocytes et est alors dégradée en sérine. Et un nouveau cycle recommence : la sérine synthétisée peut alors être utilisée renvoyée aux neurones, et ainsi de suite.
Il en est de même pour le glutamate et le GABA. Ces deux neurotransmetteurs sont recapturés par les astrocytes, qui les dégradent en glutamine. Cette glutamine est alors renvoyée aux neurones, qui pourront re-synthétiser du glutamate et/ou du GABA et les utiliser comme neurotransmetteurs. Il faut noter que le glutamate peut aussi être synthétisé par les astrocytes et les neurones non pas à partir de glutamine, mais à partir d'une molécule produite par le cycle de Krebs (respiration aérobie).

Les vitamines et le cerveau
Dans les sections précédentes du chapitre, nous avons surtout parlé de l'alimentation en énergie du cerveau. Mais il ne faut pas oublier que de nombreuses réactions chimiques permettent d'utiliser cette énergie et de la stocker. De plus, certaines réactions métaboliques permettent de synthétiser des neurotransmetteurs, de façonner la paroi des neurones, de contrôler les différences de concentrations en ions dans le neurone, et ainsi de suite. Ces réactions font naturellement appel à de nombreuses enzymes et autres protéines, la plupart étant synthétisées à partir des nutriments, mais aussi à partir de vitamines. Le cerveau est de loin un des plus gros consommateur de vitamines du corps.
D'ailleurs, il faut signaler que le cerveau est naturellement riche en vitamines, celui-ci ayant des réserves assez importantes. C'est notamment le cas pour les vitamines B, qui sont séquestrées dans le cerveau. Aussi, il n'est pas étonnant que leur teneur cérébrale soit largement supérieure à leur concentration sanguine. Par exemple, la concentration en vitamine B9 est près de 4 fois supérieure à la concentration sanguine, de même que la concentration en vitamine B5 et B8 est près de 50 fois plus importante dans le cerveau que dans le sang. Grâce à cela, le cerveau peut survivre à une déficience assez légère et/ou temporaire en vitamines. Cependant, cela n’empêche pas des déficiences en vitamines dans des situations extrêmes, comme un alcoolisme chronique ou un jeune prolongé. Ces déficiences peuvent avoir des conséquences neuropsychiatriques assez importantes, comme nous allons le voir maintenant. Dans ce qui va suivre, nous allons étudier les quatre vitamines les plus importantes pour le cerveau, à savoir les vitamines B1, B6, B9 et B12.
Les vitamines B9 et B12
Les vitamines B9 et B12 fonctionnent en tandem dans le corps, un manque de vitamine B12 pouvant être masqué par une complémentation de B9 et réciproquement. Ces cas sont cependant rares, vu que la carence en vitamine B12 ou B9 est exceptionnelle de nos jours. Elle ne touche que les végétariens qui ne se complémentent pas en vitamine B12 (et encore, après quelques mois ou années de ce régime sans complémentation) ou les personnes ayant des problèmes d'absorption des vitamines (anémie de Biermer, usage d'antiacides prolongé, problèmes intestinaux…). La récupération est souvent totale après complémentation en B12 ou B9, mais quelques patients peuvent avoir des séquelles s'ils ne sont pas pris en charge rapidement.
La vitamine B9, aussi appelée acide folique, a un métabolisme assez compliqué et est impliquée dans la réplication de l'ADN ou le métabolisme de certains acides aminés. La voie métabolique qui nous intéresse est cependant assez simple : l'acide folique est absorbé par les intestins, puis est transformé par le foie en acide lévoméfolique (5-méthyl-THF), la forme de vitamine B9 qui est absorbée par le cerveau. Ce métabolite peut traverser la barrière hémato-encéphalique en passant à travers un récepteur spécifique : le récepteur folate-alpha. Des déficits au niveau de ce récepteur peuvent entraîner une déficience cérébrale en folate dans laquelle seul le cerveau subit un déficit en B9, alors que le reste du corps a une alimentation vitaminique normale. Les symptômes sont une hypotonie, des crises épileptiques et un retard mental/psychomoteur. Sa cause principale est une maladie génétique très rare, liée à une mutation du gène FOLR1, le gène qui code pour le récepteur cérébral du folate. Une autre cause est la présence dans le sang d'anticorps qui visent ce récepteur, ce qui est une maladie auto-immune extrêmement rare.
La carence en vitamine B12 entraîne un syndrome neuro-anémique, dont le nom traduit ses symptômes : une anémie et des symptômes neuropsychiatriques variés. Pour ce qui est des manifestations psychiatriques, les symptômes vont de symptômes mineurs (troubles du sommeil, dépression) à des symptômes plus graves pouvant aller jusqu’à la démence ou des crises psychotiques. Le symptôme neurologique le plus grave est une démyélinisation du système nerveux dont les symptômes sont semblables à la sclérose en plaque, qui peut toucher la moelle épinière, les nerfs et/ou le cerveau. L'atteinte des nerfs périphériques fait que ceux-ci perdent leur gaine de myéline, donnant des polynévrites assez marquées. L'atteinte médullaire touche essentiellement le faisceau pyramidal ainsi que les cordons antérieurs (système des colonnes dorsales). L’atteinte du faisceau pyramidal et des nerfs moteurs se traduit par l'apparition de troubles moteurs tels qu’une paralysie, une spasticité, un défaut de coordination motrice ou autre. L'atteinte des nerfs sensitifs et des colonnes dorsales médullaires est la cause de troubles sensitifs variés : douleurs dans les extrémités, picotements, sensations de décharges électriques, et cætera.
La vitamine B6
Nous avons vu il y a quelques chapitres que la vitamine B6 est primordiale dans la synthèse de la sérotonine et de la dopamine, du GABA et de la mélatonine. Pour rappel, la vitamine B6 est une coenzyme associée à plusieurs enzymes. Elle active l'enzyme dopa-décarboxylase, qui dégrade le 5-HT est transformé en sérotonine et la L-DOPA en dopamine. C'est aussi la coenzyme de la GAD, qui dégrade le glutamate en GABA.
La vitamine B6 existe sous plusieurs formes différentes, mais seules trois d'entre elles entrent dans le cerveau : le pyridoxal, la pyridoxine et la pyridoxamine. Ce sont des formes inactives, qui demandent à être phosphatées pour devenir des formes actives de la vitamine B6, à savoir du phosphate de pyridoxal, du phosphate de pyridoxine ou du phosphate de pyridoxamine. Dans le cerveau, les formes inactives de B6 sont transformées en phosphate de pyridoxal par deux enzymes : la PK et la PNPO (Pyridoxine 5'-phosphate oxidase). Le phosphate de pyridoxal intervient ensuite dans la dégradation de la lysine et de la proline (deux acides aminés), via deux voies métaboliques différentes dans laquelle de nombreuses enzymes interviennent. Dans ce réseau métabolique, divers troubles peuvent survenir.
La plus fréquente, la carence en vitamine B6 perturbe le fonctionnement global du cerveau. Une déficience en vitamine B6 entraine donc une baisse de production des neurotransmetteurs cités plus haut, qui touche préférentiellement la synthèse du GABA et de la sérotonine. La baisse de GABA induite se traduit par une hausse de l'activité électrique des neurones, avec deux conséquences principales : un mauvais sommeil et, plus rarement, des crises épileptiques. La baisse de sérotonine et de dopamine se manifeste quant à elle par un état anxio-dépressif et une hausse de l'impulsivité et de la nervosité. La déficience peut aussi se manifester dans le système nerveux périphérique par une inflammation généralisée des nerfs (une polynévrite). À l'inverse, une surdose de vitamine B6 n'entraine pas de symptômes clairs, tant que la surdose n'est pas prolongée. Cependant, une complémentation en B6 prolongée durant plusieurs mois peut engendrer des névrites totalement réversibles avec l'arrêt de la supplémentation.
L'épilepsie sensible au phosphate de pyridoxal est une maladie génétique à transmission autosomique dominante. Elle est causée par une déficience en PNPO, qui touche la synthèse du phosphate de pyrixodal à partir des formes inactives de B6. Avec cette maladie, l'enzyme PNPO n'est pas synthétisée, ce qui fait que la vitamine B6 n'agit plus du tout dans le cerveau. Cela entraine une carence massive en B6 active dans le cerveau. En conséquence, la synthèse des neurotransmetteurs est perturbée et le métabolisme neuronal des acides aminés est perturbé. Le résultat est une encéphalopathie épileptique néonatale, qui survient dès les premières heures de vie du bébé atteint. Elle ne réagit pas aux traitements habituels et n'est soignée que par un traitement à base de phosphate de pyridoxine/pyridoxal. D'autres symptômes peuvent survenir, comme une hypotonie, des troubles respiratoires, des mouvements anormaux, etc.
Similaire à la maladie précédente, l'épilepsie pyridoxine-dépendante est un ensemble de maladies génétiques caractérisées par une encéphalopathie néonatale similaire à la maladie précédente. Les crises épileptiques sont de type myocloniques, avec un tracé typique sur l'EEG. La seule différence est que la maladie est sensible à un traitement à base de pyridoxine, une forme inactive de la B6, là où la précédente a besoin de la forme active de la B6. L'identification de cette maladie est souvent difficile à faire et le diagnostic se fait après administration de vitamine B6 ou un diagnostic génétique. Elle se manifeste très tôt : dans les premiers jours ou mois de vie pour les formes précoces, vers 1 à 3 ans pour les formes tardives. Ce syndrome touche entre une naissance sur 500 000 et une naissance sur 400 000. Ses causes sont nombreuses et de nombreux mutations génétiques peuvent la causer. Dans les grandes lignes, il existe deux sous-syndromes principaux : une forme précoce qui apparait dans les premières heures de vie, et une forme tardive qui apparait vers 1 à 3 ans.
- La forme précoce exprime une épilepsie prénatale dès 20 semaines de gestation. L'épilepsie se manifeste rapidement et toutes les formes de crises épileptiques peuvent survenir : myoclonies, absences, crises toniques, toniques-cloniques. Elle est souvent secondée par de l'irritabilité et une réaction excessive aux stimuli, ainsi que par des malformations cérébrales diverses (hydrocéphalie, malformations du corps calleux, ...). On peut observer des troubles métaboliques généraux, des troubles respiratoires, et bien d'autres symptômes divers. Sans traitements, la maladie entraine un retard mental et divers déficits neurologique et développementaux. La forme précoce la plus fréquente est causée par une mutation qui perturbe la synthèse de l'antiquitine, une enzyme de la voie de dégradation de la lysine.
- Par contraste, la forme tardive apparait avant 3 ans et répond initialement aux médicaments anti-épileptiques, avant que ces traitements cessent de faire effet. L'épilepsie est isolée, avec peu d'autres symptômes neurologiques et une absence de malformations cérébrales.
La vitamine B1
La vitamine B1, aussi appelée thiamine, est une vitamine du fameux cycle de Krebs (une série de réactions chimiques qui fournit de l'énergie aux cellules vivantes). Une carence en vitamine B1 entraine une pénurie d'énergie et d'ATP dans les cellules, qui se mettent à dysfonctionner et parfois à mourir. Le système nerveux étant un tissu très gourmand d'un point de vue métabolique, toute carence en vitamine B1 retenti en premier lieu sur le fonctionnement cérébral. Autant dire qu'elle est extrêmement importante pour le système nerveux, des carences pouvant être tout aussi graves que les carences en vitamines B12 ou B6. La carence en thiamine cause un stress métabolique aux neurones, qui cause des dysfonctionnements divers des potentiels d'action, mais qui peut aussi entrainer souvent la mort du neurone par apoptose. De plus, la carence va aussi modifier la recapture du glutamate, entrainant l'apparition d'une excitotoxicité (le glutamate excite les neurones à mort). Beaucoup de neurones dysfonctionnent, quand ils ne meurent rapidement, ce qui cause l'apparition d'une maladie : l'encéphalopathie de Wernicke.
Ses symptômes sont souvent assez clairs, 80% des cas manifestant la triade : défaut de coordination des mouvements (ataxie), paralysie des yeux (ophtalmoplégie) et confusion (delirium). D'autres symptômes peuvent se faire jour, comme une profonde amnésie, une perte de la mémoire à court-terme, une psychose, ou des troubles végétatifs. L'origine de ces symptômes est liée à diverses atteintes du thalamus, de l'hypothalamus et du tronc cérébral. Par exemple, la paralysie oculaire provient d'une atteinte des noyaux des nerfs crâniens oculomoteurs. L"amnésie est quant à elle liée à une atteinte des corps mamillaires de l'hypothalamus. Une carence prolongée en B1 s'observe surtout chez les alcooliques, l'alcool stoppant l'absorption intestinale de la B1.
Chez certains patients, l'encéphalopathie de Wernicke évolue vers des troubles cognitifs permanents. La démence qui en résulte est appelée le syndrome de Korsakoff, du nom de son découvreur. Son symptôme principal est une amnésie assez particulière. Nous allons devoir parler rapidement de l'amnésie, chose qui sera détaillé dans le chapitre sur la mémoire. L'amnésie du syndrome de Krosakoff est une perte de la mémoire à court-terme, avec une incapacité à mémoriser de nouveaux souvenirs ou de nouvelles connaissances. Les savoirs et souvenirs déjà acquis sont relativement préservés, bien que quelques déficits peuvent se voir. L'amnésie porte donc essentiellement sur les acquisitions qui suivent le traumatisme, cette forme d'amnésie étant appelée amnésie antérograde. L'amnésie qui touche les souvenirs d'avant l'apparition du syndrome est appelée amnésie rétrograde. L'amnésie du syndrome de Korsakoff est essentiellement antérograde, bien qu'une faible amnésie rétrograde soit possible. Si amnésie rétrograde il y a, celle-ci ne touche généralement que les souvenirs et savoirs récents, qui datent de quelques mois, années ou décennies avant le syndrome. Outre l'amnésie, divers troubles cognitifs peuvent se manifester, que ce soit des troubles du langage (aphasie), des troubles de la reconnaissance des objets et de la catégorisation (agnosie) ou des troubles intellectuels.
Les minéraux et le cerveau
Outre les vitamines, les mal-nommés « minéraux » sont d'une importance capitale pour le fonctionnement normal du cerveau. Les « minéraux » les plus importants sont de loin les ions Calcium, Potassium et Sodium, sans lesquels il ne peut y avoir de potentiels d'action. Leur rôle a déjà été abordé dans le chapitre sur potentiel d'action et nous n'en reparlerons pas ici. Les autres ions que nous allons aborder sont le magnésium, le cuivre, le fer et quelques autres. Vous remarquerez qu'il s'agit d'ions métalliques, à quelques exceptions près.
Tous les ions ont une concentration particulièrement bien régulée, le cerveau disposant de toute une machinerie chimique pour régler leur concentration. Il faut dire que ces ions deviennent toxiques quand ils sont en grandes quantités. C'est notamment le cas des ions métalliques, qui sont impliqués dans des réactions d’oxydoréduction qui créent des molécules « poisons » (des radicaux libres). En temps normal, les produits nocifs de ces réactions sont éliminés ou dégradés par la machinerie cellulaire du cerveau, mais ces processus sont dépassés quand la concentration en ions métalliques devient trop importante. De même, une déficience est nuisible pour les neurones, ces ions servant dans de nombreuses réactions chimiques importantes pour le fonctionnement des neurones.
La concentration en ions du cerveau est sévèrement contrôlée par divers mécanismes, dans lesquels la barrière hémato-encéphalique joue un rôle crucial. Elle protège le cerveau des variations de concentration ionique du sang. Par exemple, prenons le cas où la concentration en sodium du sang augmente, suite à la consommation d'un aliment particulier, d'une réponse hormonale, ou d'un supplément alimentaire. Le cerveau ne va pas être impacté par cette variation, et en sera isolé : la concentration ionique intracérébrale restera la même. C'est grâce à la barrière hémato-encéphalique, qui isole le cerveau des vaisseaux sanguins. L'absorption d'ions dans le cerveau, ou leur excrétion, est réalisée par des canaux ioniques et/ou des transporteurs, qui se trouvent à la surface de la barrière hématoencéphalique. Elles vont se lier aux minéraux ou aux vitamines à absorber et vont les rapatrier dans les astrocytes pour l'absorption. Une partie des ions est perdue dans le liquide céphalorachidien, et est emportée avec lui lors de son excrétion dans le sang.
Le magnésium
L'ion magnésium a aussi été vu dans le chapitre sur les récepteurs synaptiques et la plasticité synaptique. Nous avons vu que les récepteurs NMDA du glutamate sont bloqués par un ion magnésium, qui bouche le canal ionique. Du moins, c'est le cas tant que le potentiel du neurone reste inférieur à -60 mV. Si la tension dépasse ce seuil, l'ion Magnésium est éjecté et le canal ionique s'ouvre, excitant le neurone. On devine donc qu'un manque en magnésium se traduit par une hyper-excitabilité neuronale, alors qu'un excès de Magnésium entraine une hypo-excitabilité neuronale. Dans les deux cas, l'atteinte neuronale est diffuse, mais ciblée sur les neurones sensibles au glutamate.
- La déficience en magnésium entraine des symptômes neurologiques qui vont d'une simple asthénie et/ou une faiblesse musculaire, à des symptômes plus sérieux comme des convulsions et plus rarement un coma.
- Pour un excès en magnésium, les symptômes neurologiques sont frustres pour les faibles hypermagnésémies, mais bien plus lourds dans les cas graves. À faible dose en excès, le magnésium entraine une simple léthargie et/ou asthénie. Pour un excès assez fort, elle surtout des symptômes neuromusculaires. Il a, à hautes doses, un effet sur la jonction neuromusculaire similaire à celui du curare. En clair, elle cause une décontraction musculaire, une diminution des réflexes, des paralysies (notamment respiratoires). Dans certains cas extrêmes, on observe une dilatation pupillaire liée à un blocage du système nerveux sympathique.
Le cuivre
Le cuivre n'a pas de rôle physiologique clair, y compris au niveau du cerveau. Tout ce que l'on sait est que des excès ou des carences entrainent de nombreux symptômes au niveau du foie, du cerveau et d'autres tissus/organes. Le cuivre est absorbé par l'intestin, passe par le foie et atteint le cerveau. Divers transporteurs permettent au cuivre de traverser la barrière intestinale et la barrière hémato-encéphalique. Chez certaines personnes, ces transporteurs dysfonctionnent, ce qui fait que le cuivre ne rentre pas, ou au contraire s'accumule, dans les tissus. Les maladies liées à l'accumulation de cuivre dans le cerveau se manifestent (entre autres) par des symptômes neurologiques assez francs. Toutes touchent toutes les ganglions de la base, ce qui entraine des symptômes caractéristiques : parkinsonisme, mouvements involontaires anormaux (chorée, tics, dyskinésies, autres), troubles psychiatriques variés, atteinte cognitive.
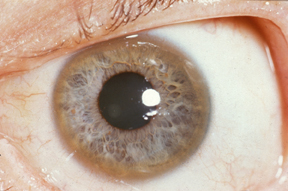
La maladie de Wilson est une maladie (en fait, un ensemble de maladies génétiques) qui entraine une accumulation de cuivre dans l'organisme. Les organes les plus touchés sont le foie et le cerveau. Seuls une moitié des patients manifestent ces symptômes neuropsychiatriques. Les autres symptômes sont des problèmes de foie et aux yeux, parfois au cœur et aux reins. Les patients ont presque tous des troubles au foie, notamment une cirrhose, qui peuvent cependant passer inaperçus quand leurs symptômes sont frustres (ictère, fatigue, saignements, hypertension dans la veine porte, ...). Chez certains patients, on observe un anneau brun sur le pourtour des iris et des yeux, quasi-pathognomonique de la maladie. Au niveau cérébral, l'accumulation de cuivre est toxique pour les neurones, qui meurent progressivement. Les symptômes de la maladie traduisent une atteinte des ganglions de la base, qui sont les aires cérébrales les plus touchées par l'accumulation de cuivre.
- Les symptômes neurologiques sont des problèmes d'équilibre (ataxie), un parkinsonisme, des troubles du tonus musculaire (dystonie). Par contre, les troubles sensoriels sont très rares, au point que leur présence signifie pour le médecin qu'il vaut mieux chercher un autre diagnostic. Plus rarement, une épilepsie peut survenir, mais ce n'est pas un symptôme diagnostic.
- Les symptômes psychiatriques sont assez variés, mais sont essentiellement des troubles du comportement : désinhibition, irritabilité extrême, impulsivité, etc. On observe aussi fréquemment des troubles l'humeur (dépression), de l'anxiété. On observe plus rarement une psychose (syndrome regroupant hallucinations, délires, désorganisation de la pensée et troubles du comportement). Il arrive que les patients subissent des troubles cognitifs/intellectuels, qui miment une démence : pertes de mémoire, des troubles de l'idéation (pensée ralentie), et autres.
Si l'excès de cuivre dans le cerveau entraine des maladies, la déficience en cuivre aussi. La maladie de Menkes est causée par un dysfonctionnement des transporteurs du cuivre, ce qui fait que le cuivre ne pénètre plus dans les tissus. Elle entraine une déficience en cuivre, qui touche notamment le cerveau. Les neurones meurent rapidement, la neurodégénération touchant tout le cerveau. De plus, la myéline ne se forme pas autour des axones. Les patients montrent un manque de tonus musculaire (hypotonie), de l'épilepsie et divers symptômes neurologiques variés. On observe aussi un visage déformé, caractéristique de la maladie, une perte des cheveux, et quelques autres symptômes. Il s'agit d'une maladie grave qui apparait quelques mois après la naissance. Les enfants touchés ne dépassent pas l'âge de 3-5 ans.
Le fer
Le fer est utilisé par tous les tissus et toutes les cellules. L'utilisation la plus courante du fer est celle des globules rouges, dont l'hémoglobine est composée en partie de fer. C'est d'ailleurs au fer de l’hème que l’oxygène et le dioxyde de carbone se fixent sur l'hémoglobine. Mais l'utilisation la plus importante par les cellules est la fabrication d'énergie. Les cellules se servent du Fer pour fabriquer de l'énergie sous forme d'ATP. Pour être précis, il sert pour la respiration cellulaire, au niveau de la chaine de transport d'électrons des mitochondries (ce détail aura son importance pour l'ataxie de Friedrich). Et comme toutes les autres cellules, les neurones ont besoin de fer pour fabriquer de l'ATP et survivre.
Le fer est absorbé au niveau de l'intestin, grâce à l'action d'au moins un transporteur. Une fois absorbé, le fer est libéré dans le sang et se lie à une molécule de transport : la transferrine. La transferrine, transportée par le sang, relâche le fer au niveau des tissus qui en ont besoin. L'absorption du fer par l'intestin est assez mauvaise, surtout pour le fer végétal, mais est améliorée en présence de vitamine C. Pour compenser sa faible absorption, l'excrétion du fer est très faible, au point que le fer est quasiment séquestré dans l'organisme une fois absorbé. Mais cela n’empêche pas le corps de gérer au mieux les maigres réserves en fer, pour éviter toute pénurie. Il est parfois stocké au niveau du foie sous la forme de ferritine, une molécule de stockage du fer dans l'organisme. Ce système de stockage hépatique du fer permet à celui-ci de garder constante la teneur en fer du sang : il relâche du fer en cas de manque et en stocke en cas d'excès. De plus, le fer présent dans les cellules est systématiquement recyclé quand elles meurent. En effet, les macrophages, qui avalent et digèrent les cellules mortes, relâchent dans le sang le fer des cellules ainsi phagocytées. Tout cela fait que la carence martiale (la carence en fer) est rare avec une alimentation équilibrée, seules quelques maladies métaboliques ou génétique pouvant causer des manques de fer. L'excès en fer est aussi très rare, l'absorption et le stockage du Fer étant régulé par diverses hormones comme l'hepcidine.
Quoi qu’il en soit, le fer sanguin,lié à la transferrine finit naturellement par arriver au cerveau. Mais pour cela, il doit traverser la barrière hémato-encéphalique, qui régule son entrée dans le système nerveux. Le fer se détache de la transferrine et se fixe sur des transporteurs présents à la surface de la barrière hémato-encéphalique. Le fer traverse alors la barrière et se retrouve dans le cerveau (dans les astrocytes, puis dans les neurones). Précisons que le fer capté par ce transporteur est obligatoirement un ion et non les autres formes ioniques ( ou ). Une fois entré dans le cerveau, le est immédiatement oxydé en par les astrocytes, avant de se lier à une transferrine intracérébrale. La transferrine se lie à divers récepteurs à la surface des oligodendrocytes et des neurones cérébraux : le fer se détache de la transferrine et rentre dans ces cellules. Le fer s'accumule préférentiellement dans les ganglions de la base et la substance noire (la substantia nigra pars compacta, pour être précis).
Précisons que la transferrine cérébrale est plus efficace que la transferrine sanguine. Là où la transferrine sanguine est saturée à seulement 30% de sa capacité de transport maximale, la transferrine cérébrale atteint presque 100%. Il s'agit pourtant de la même molécule, mais les différences chimiques entre le sang et le tissu cérébral font que... Une conséquence est la transferrine ne peut pas servir de tampon en cas d'excès en fer cérébral, vu qu'elle est déjà chargée au maximum, ce qui rend les neurones très sensibles à une surcharge en fer.
Il arrive que le cerveau se retrouve soit en manque chronique de fer, soit qu'il est surchargé de fer au point où les teneurs atteintes sont toxiques. Les maladies responsables sont des maladies génétiques très rares, regroupées sous le nom de Neurodegeneration with brain iron accumulation, abrévié NBIA. L'accumulation touche en priorité les ganglions de la base et le cervelet, à savoir les aires cérébrales les plus chargées en fer. Elle se traduit donc par une symptomatologie caractéristique d'une atteinte des ganglions de la base, à savoir un syndrome parkinsonien ou d'autres mouvements anormaux (chorée, dystonies), parfois couplés à un déclin intellectuel/cognitif. Les mécanismes de l'accumulation du fer dans le cerveau sont multiples et peuvent toucher des molécules très diverses. Les médecins ont identifié une petite dizaine de sous-types de NBIA, chacun se distinguant des autres par le gène muté et les mécanismes de l'accumulation du fer.
| Type de NBIA | Gène muté |
|---|---|
| Neurodégénération associée à la panthotenate-kinase (PKAN) | PANK2 |
| Neurodégénération associée à PLA2G6 (PLAN) | PLA2G6 |
| Neurodegeneration associée aux protéines de la membrane mitochondriale (MPAN) | C19orf12 |
| Neurodégénération associée à la protéine Beta-Propeller (BPAN) | WDR45 |
| FAHN | FA2H |
| Syndrome de Kufor-Rakeb | ATP13A2 |
| Neuroferritinopathy | FTL |
| Acéruloplasminémie | CP |
| Syndrome de Woodhouse-Sakati | DCAF17 |
| CoPAN | COASY |
- Pour ceux qui veulent en savoir plus sur les NBIA, je conseille la lecture de ce lien : NBIA disorders association
Mais il existe des maladies qui sont causées non pas par une accumulation de Fer dans le cerveau, mais par des dysfonctionnements plus compliqués à expliquer. L'ataxie de Friedreich est une de ces maladies. Cette maladie est causée par une mutation qui perturbe la fabrication d'une enzyme, la frataxine. Le métabolisme intracellulaire du Fer est perturbé, et plus précisément le métabolisme mitochondrial. Le résultat est que les cellules des tissus fortement consommateurs d'ATP meurent rapidement, les neurones ne faisant pas exception. De plus, la gaine de myéline des neurones se dégrade et finit par disparaitre, sans être remplacée. La maladie démarre aux alentours de l'adolescence et se manifeste principalement par des problèmes d'équilibre et de coordination des mouvements (une ataxie, qui donne son nom à la maladie). Par la suite, on observe des troubles neurologiques moteurs et sensoriels, assez divers, d'apparition progressive. Certains sont causés par une atteinte du cervelet (troubles de l’articulation, des mouvements oculaires), par une atteinte faisceau pyramidal (paralysie, faiblesse musculaire), et/ou par une atteinte de la moelle épinière. Typiquement, le patient a du mal à marcher, perd facilement l'équilibre, a du mal à coordonner ses mouvements. Puis, le patient perd progressivement l'usage de ses bras et de ses jambes, il ressent une faiblesse musculaire envahissante. Parfois, il a du mal à articuler, ses mouvements oculaires sont erratiques. Quand sa moelle épinière et atteinte, sa sensibilité corporelle se dégrade, son sens du toucher et sa proprioception disparaissent. Beaucoup plus rarement, les nerfs optiques et auditifs se démyélinisent, causant perte d'acuité visuelle ou auditive, cécité, surdité, etc. Précisons pour finir que la maladie ne touche pas que le cerveau, mais cause aussi des atteintes cardiaques (très fréquentes), une scoliose ou d'autres déformations osseuses, et parfois un diabète (10-20% des patients).
Pour ceux qui veulent en savoir plus sur le métabolisme cérébral du fer, je conseille la lecture de cet article scientifique, en libre accès sur pubmed :