get_support function does not display branch supports for all #1887
Comments
|
I have edited my issue to add images to explain the issue more clearly. |
|
Where is |
|
Yeah sure, I can provide the sequence text file in FASTA format. The and here is the |
|
Thanks. Have you looked to see if all the bootstrap values are there if if saved to a tree file (e.g. Newick format, or PhyloXML)? Either look at the files, or try opening them in a tool like FigTree. This should tell you if the problem is in the data, or the drawing. |
|
Alright, I am trying to check it this way. I will tell you once the results are generated. The size of the window remains fixed no matter how larger the data becomes for display. Are'nt there any way for making the window size scrollable according to data file size , so that it gives a good display? |
|
I think you would need to look at enhancing the I practice I would use the Biopython code for large scale automation (e.g. generating hundreds of trees and images), but when I wanted to prepare a single image for publication I would like use a dedicated package like http://tree.bio.ed.ac.uk/software/figtree/ (which can help with things like colour coding). |
|
Can't the axes sizes be made flexible for |
|
Yes in principle, although it is less clear how best to extend the API for flexibility and ease of use (more and more optional arguments for things like image size and font size could become unwieldy). If you worked on this you'd be in a position to suggest something, but it would down to @etal to review it. |
|
The things is I can't do like, first generating a tree and then go to some tool to display it. This is the reason I wanted your help to suggest me how can I enhance the window axes for display so that it becomes scrollable if the file data size is larger. |
|
One thing that I do not understand is that, when I use the first tree from bootstrap replicates to be compared with the rest of the replicates , the resulted tree shows a high confidence values for all of the branches, even when I used just 50 bootstrap value. |
|
Your examples are still incomplete, you are missing the imports - this is enough for the second example to run: The first example does not define |
|
No , see my imports. If I have'nt been using them, I could not have generated the tree files. I am really sorry that I forgot to mention. |
|
Here is the complete code; This is how an MSA data file is being ued to generate a tree and then bootstrap replicates for this data are produced. Then using |
|
Adding this line to your complete example is instructive: Phylo.write(support_tree, "example.bootstrap1200.xml", "phyloxml")Quoting in full, <phyloxml xmlns="http://www.phyloxml.org" xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" xsi:schemaLocation="http://www.phyloxml.org http://www.phyloxml.org/1.10/phyloxml.xsd">
<phylogeny rooted="true">
<clade>
<branch_length>0</branch_length>
<confidence type="unknown">100.0</confidence>
<clade>
<name>Amphioxus</name>
<branch_length>0.606194690265</branch_length>
</clade>
<clade>
<name>Inner2</name>
<branch_length>0.163337565699</branch_length>
<clade>
<name>Inner5</name>
<branch_length>0.0357208702065</branch_length>
<clade>
<name>Inner7</name>
<branch_length>0.0603068153884</branch_length>
<clade>
<name>Inner8</name>
<branch_length>0.0112301806785</branch_length>
<clade>
<name>Inner4</name>
<branch_length>0.0579106089338</branch_length>
<confidence type="unknown">100.0</confidence>
<clade>
<name>Chicken</name>
<branch_length>0.0592223861029</branch_length>
</clade>
<clade>
<name>Zebra</name>
<branch_length>0.0882702392658</branch_length>
</clade>
</clade>
<clade>
<name>Inner6</name>
<branch_length>0.0260808443953</branch_length>
<clade>
<name>Inner10</name>
<branch_length>0.0481348328417</branch_length>
<confidence type="unknown">0.166666666667</confidence>
<clade>
<name>Inner12</name>
<branch_length>0.0106845731932</branch_length>
<confidence type="unknown">0.5</confidence>
<clade>
<name>Inner9</name>
<branch_length>0.0152994791667</branch_length>
<confidence type="unknown">95.75</confidence>
<clade>
<name>Dog</name>
<branch_length>0.0476585545723</branch_length>
</clade>
<clade>
<name>Cow</name>
<branch_length>0.0585361356932</branch_length>
</clade>
</clade>
<clade>
<name>Inner11</name>
<branch_length>0.00483095962389</branch_length>
<confidence type="unknown">1.75</confidence>
<clade>
<name>Inner3</name>
<branch_length>0.0761459485619</branch_length>
<confidence type="unknown">100.0</confidence>
<clade>
<name>Human</name>
<branch_length>0.00359513274336</branch_length>
</clade>
<clade>
<name>Chimpanzee</name>
<branch_length>0.00525442477876</branch_length>
</clade>
</clade>
<clade>
<name>Mouse</name>
<branch_length>0.119096454031</branch_length>
</clade>
</clade>
</clade>
<clade>
<name>Elephant</name>
<branch_length>0.0678091583702</branch_length>
</clade>
</clade>
<clade>
<name>Platypus</name>
<branch_length>0.128667562158</branch_length>
</clade>
</clade>
</clade>
<clade>
<name>Anole</name>
<branch_length>0.154110588741</branch_length>
</clade>
</clade>
<clade>
<name>Xenopus</name>
<branch_length>0.202110988201</branch_length>
</clade>
</clade>
<clade>
<name>Inner1</name>
<branch_length>0.0294985250737</branch_length>
<confidence type="unknown">99.25</confidence>
<clade>
<name>Fugu</name>
<branch_length>0.213864306785</branch_length>
</clade>
<clade>
<name>Zebrafish</name>
<branch_length>0.213864306785</branch_length>
</clade>
</clade>
</clade>
</clade>
</phylogeny>
</phyloxml>Some but not all of the inner nodes (which have been automatically named) do seem to have confidences, such as The question becomes are these bootstrap values really missing, or are they really branches with zero boot strap support? Given the way Python evaluates numbers when cast as a boolean, a bootstrap value of zero is a special case and that might be why it was not reported in the XML output, or the drawing: https://github.com/biopython/biopython/blob/biopython-173/Bio/Phylo/_utils.py#L370 |
|
So what does it conclude? The particular branching points have 0 confidence ? And if it does so, how do I change it so that it may show the value whether zero? Do you get my point of confusion? |
|
My guess right now is the missing bootstrap values are simply zero. Do you agree that the same values are missing in the figures as in the data files? For example, look at the saved trees in a text editor or graphical tree tool. If so, that is good progress - it strongly suggests the problem is not in the tree drawing. If you do not want to delve into debugging Biopython, and need to resolve this urgently, try doing the bootstrap analysis in another set of tools - for example PHYLIP. It used to be good advice to generate phylogenetic trees by at least two different methods anyway, to confirm they give reasonably consistent results, and so provide reassurance that your analysis is not based on an artefact of a particular tree building tool. |
|
Alright, I might test my trees again. |
|
Ain't there any way for displaying '0' for the branch support where it is zero rather showing nothing? |
|
If you want to experiment with the non-display of zero bootstraps, try changing this line: https://github.com/biopython/biopython/blob/biopython-173/Bio/Phylo/_utils.py#L370 Currently: if clade.confidence:
return conf2str(clade.confidence)This might work better by showing zero: if clade.confidence is not None:
return conf2str(clade.confidence)I am guessing that |
|
I think |
|
Yeah okay, I 'll give it a try now. |
|
@etal I would expect the line of code to be exercised by the existing tests, and with graphical output we don't currently have anyway of confirming the output except inspection by eye - so sure, if it works in a hands on test, go ahead and commit it. Is there potentially something similar happening with the PhyloXML (and other format) output? |
|
|
Ommission found while exploring #1887, fix agreed by Eric Talevich.
|
I tested adding (I did a quick search and replace of Given @etal's comment earlier, I went ahead and committed that fix in 5ba7092 to label zero value bootstraps in However, this alone does not solve @SidraYounas's problem. Someone needs to dig into the data structure to understand why these interior nodes don't seem to have a bootstrap value. |
Setup
I am reporting a problem with Biopython version, Python version, and operating
system as follows:
Expected behavior
I have used the
get_support()function from Biopython.Phylo module to generate the support values for my original tree branches as follows:Example sequence file is:

example.pdf
##The required result tree should be like: (i.e, it shows branch support values for every node point.)
The behavior of module being faced
While the
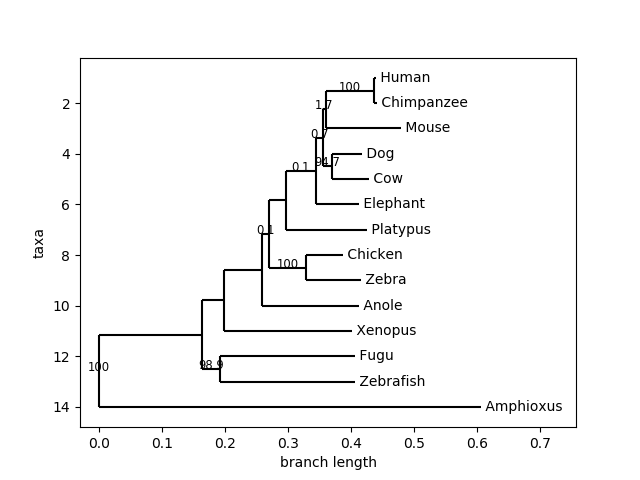
Phylo.draw(support_tree)is executed, the resulted tree shows values on a few of the branches and not for all of the branching points, (as the tree image shown)The resulting tree is:
Can you suggest any solution for this?
one more thing: If I generate a bootstrap consensus tree for the same MSA data, as follows:
it does show the bootstrap support values for all of the branchng points as:

and none of the value is in floating number rather integers while they are shown as floating numbers for my tree.
The text was updated successfully, but these errors were encountered: